Several IGB users asked us to add the latest fruit fly genome and annotations to IGB. Here I describe how I did this, using data files downloaded from FlyBase, the chief annotation authority for fruit fly genomes.
In July 2014, FlyBase and Berkeley Drosophila Genome Project (BDGP) released a new genome assembly for the fruit fly genome Drosophila melanogaster. They also released a new set of gene model annotations in various formats – including GFF, GTF, and XML.
To make the data available to IGB users, I needed to download the sequence data and reference gene model annotations, convert them to formats IGB can use for visualization, and then deploy the converted files on the IGBQuickLoad.org Web site. This is the Web site that IGB uses to retrieve data for annotated, sequenced genomes. When IGB users select a genome, the reference gene model track data that automatically loads into IGB comes from the IGBQuickLoad site or one of its mirror sites.
Adding a new genome directory to the IGBQuickLoad data repository
To start, I added a new genome version directory (folder) to the IGBQuickLoad.org data repository. I did this using “svn”, the program we use to manage the data sets. I used the “svn mkdir” command to create a new directory named D_melanogaster_Jul_2014, following our naming convention for genome versions. In IGB, we use the genus, species, release month and year to specify a genome version. (Sometimes we also include an extra name to indicate variety or something else, such as Z_mays_B73.) However, in this case, we don’t need a variety suffix, so we only use the genus and species to indicate the genome.
The IGBQuickLoad.org data repository is version-controlled as part of a large subversion (svn) repository – the master copy is hosted at https://svn.transvar.org/repos/genomes/trunk/pub/quickload/ and is publicly accessible. Which means: anyone who wants a copy can use “svn co” to get it.
The IGB team has been using subversion for many years to track important data files, like gene annotations or sequence, that people use for data analysis and visualization in IGB. To save effort, we also use the same repository to serve data files to IGB, using the IGB QuickLoad mechanism. When we need to deploy a new quickload site – on our local computers or via a Web server – we just use “svn co” to check out a fresh copy and then copy over some bigger data files (e.g., BAM files) that are too large to add to the repo. However, genome sequence files for smaller genomes (like fruit fly) are not very large and so I usually version-control these in the repository along with the gene model files. (More on this later.)
Adding genome sequence to the repository
Next, I looked around the FlyBase.org Web site to find a file containing the new genome assembly sequences. I found it here: ftp://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.03_FB2014_06/fasta/. The directory contained a file named dmel-all-chromosome-r6.03.fasta.gz. Based on the name, I guessed this file contained the genome sequence, so I downloaded that.
Next, I used Jim Kent’s faToTwoBit program to convert the file from fasta to 2bit format, a compressed, indexed format that enables IGB (and other programs) to retrieve data from the file without having to read the entire thing – called “random access.” This is what enables IGB to load just part of a genomic sequence, which is faster and more memory-efficient than loading the entire thing.
After converting the fasta file to 2bit, I next used Kent’s twoBitInfo program to make a genome.txt file, which is a tab-delimited file that lists the sequences and their sizes. When a user selects a genome version in IGB, IGB then sends a request to a QuickLoad site that supports that genome, asking for a copy of that genomes genome.txt file, which is stored in the genome directory for that genome version. IGB reads this file and uses the information to populate the Current Sequence tab on the right side of the main IGB window. The Current Sequence tab displays a sortable, clickable table that you can use to change chromosomes in the main display.
This is different from (and we think better than) other genome browser designs, where users have to operate a menu or click on a to-scale genome graphic to change which sequence is on display. Menus don’t work well if there are more than a few items in the menu, and a to-scale genome graphic is even worse because smaller contigs are too hard to select. That’s why in IGB, we use a sortable table instead. And, of course, users can also search for contigs and genes by name.
A quick review of the genome.txt file revealed that this new assembly contains the expected fruit fly chromosome arm sequences (e.g., 2L), but it also contains a lot of other sequences with identifiers like “211000022280328” and “2Cen_mapped_Scaffold_10_D1684.” Altogether, the genome sequence file contains 1,870 sequences.
Interesting! I was not expecting to find so many sequences in this release because the previous release (dm3) contained the autosomal chromosome arms and the sex chromosomes only. But including these additional sequences makes sense. I imagine that the assembly creators at BDGP did not want to omit some-one’s favorite gene from the release just because it didn’t assemble into the main chromosome arms.
Getting gene model annotations from FlyBase
Looking around a bit more, I found these two folders: ftp://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.03_FB2014_06/gtf/ and ftp://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.03_FB2014_06/gff/.
I reviewed the files in each folder, starting with the GFF files. I’ve never had good experiences with GFF, mainly because different people use it in difference ways, despite many attempts to standardize the format. At first glance, the FlyBase GFF looked too complex to handle easily.
So next, I turned to the files in the GTF directory. I don’t know why FlyBase included these files in their ftp site, but I’m guessing it had something to do with supporting users who want to use CuffLinks to analyze RNA-Seq data. CuffLinks uses GTF to represent gene models, andI already have code that converts CuffLinks GTF to BED-detail, our preferred format for distributing gene model data files in IGB. A quick review of the file showed me that the format was indeed simpler, and so I decided to work with that file instead of the GFF files.
Unfortunately, my Cufflinks GTF conversion code failed on the FlyBase GTF file because it lacked transcript features. Also, FlyBase included many in-line comments.
Nonetheless, it looked simpler, and so I wrote a new script to handle FlyBase GTF as distinct from other forms of GTF. The script is named flybaseGtfToBed.py and resides in bitbucket repo https://bitbucket.org/lorainelab/genomes_src. I used this new code to create a BED-detail file named D_melanogaster_Jul_2014.bed, which contains gene models converted from the GTF file.
About BED-detail and how we use it in IGB
BED-detail, also known as BED14, has all the same fields as BED12, plus two more. In IGB, we use these extra fields to provide useful meta-data about a gene model. In IGB, field 13 contains the gene name for a gene model. And field 14 contains human-friendly text describing the gene’s function. When users open these files in IGB, IGB formats them internally in a way that allows users to do keyword searches on field 14. This is very useful, but of course it depends on having human-friendly text available to use in this field. For Arabidopsis, we use the “curator summary” text in field 14. I’m not sure what we can use for fruit fly as I have not been able to find an equivalent to the Arabidopsis “curator summary.”
BED-detail benefits in other areas – high-throughput data analysis
We use BED-detail mainly because it’s compact and easy to index using tabix. (More on this later.) Also, the structure of the BED-detail files makes them extremely useful in high-throughput data analysis using R/Bioconductor tools. My favorite RNA-Seq data analysis trick to find the smallest start and largest end position for all transcripts belonging to the same gene, and I identify those by grouping on gene symbol. Then I use the coordinates to count reads overlapping the gene. Once I have those counts, I can feed them to edgeR or DESeq or other similar tools.
Deploying the new gene model BED-detail file on IGBQuickLoad
For simplicity, I named the file after the genome release: D_melanogaster_Jul_2014.bed. Next, I sorted the file by sequence name and start position, compressed it using bgzip, indexed it using tabix, and added the files to our subversion repository:
sort -k1,1 -k2,2n D_melanogaster_Jul_2014.bed | bgzip > D_melanogaster_Jul_2014.bed.gz tabix -s 1 -b 2 -e 3 D_melanogaster_Jul_2014.bed.gz svn add D_melanogaster_Jul_2014.bed.gz.* svn ci -m "Converted from FlyBase GTF using flybaseGtfToBed.py"
Adding meta-data file: annots.xml and HEADER.html
to-do
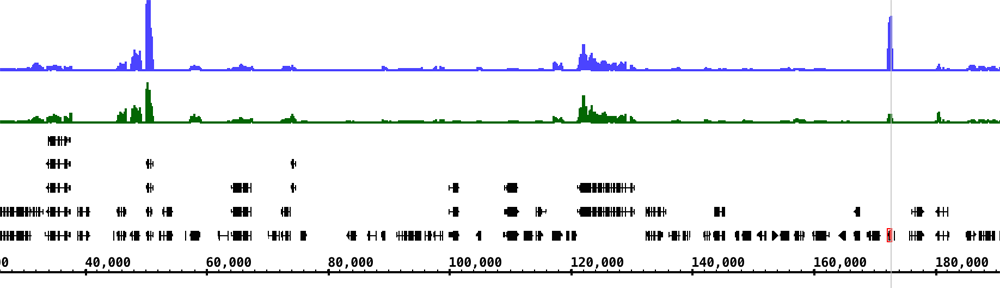
Viewing the data in IGB
Here’s an image from IGB showing an alternatively spliced gene (para) from chromosome X. The depth graph made from the gene models (FlyBase) track highlights differentially spliced regions. Places where the depth graph changes height indicate alternative splicing.
